










Regulatory, Pricing and Reimbursement Overview
Carey / Chile
A brief overview of the situation regarding regulation, pricing and reimbursement of drugs in Chile. Prepared in association with Carey, a leading global law firm, this is an extract from The Pharma Legal Handbook: Chile, available to purchase here for GBP 99.
1. What are the regulatory authorities with jurisdiction over drugs, biologicals, and medical devices in your country?
In Chile the regulatory authority responsible for the enforcement of the regulatory framework for pharmaceutical products, including biologicals, and medical devices is the Public Health Institute (ISP), which is a functionally decentralized and autonomous public service overseen by the Ministry of Health (MoH).
In turn, the Ministry of Health is the main health authority in Chile, which, pursuant to the provisions of the Chilean Sanitary Code, is responsible for the issuance of the respective regulations which govern the import, clearance, export, production, manufacturing, fractioning, storage, handling, transport, distribution, sale, pharmacovigilance, traceability, advertising, promotion or information to professionals, medical use or scientific investigation of pharmaceutical products and for the progressive implementation of the provisions for medical devices.
2. What is the regulatory framework for the authorization, pricing, and reimbursement of drugs, biologicals, and medical devices?
In Chile, the authorization for the commercialization of a pharmaceutical product is governed by the Sanitary Code, the regulations set forth in Supreme Decree No. 3/2010, issued by the MoH, which contains the Regulations for the National Control System of Pharmaceutical Products for Human Use and by ancillary regulations and technical guidelines approved by the MoH and the ISP (e.g. Decree No. 500 of 2012 of the MoH approving Technical Guideline No. 136 establishing the active ingredients included in pharmaceutical products that must demonstrate therapeutic equivalence and its amendments; Decree No. 27 of 2012 of the MoH approving Technical Guideline N° 131 defining the criteria to prove therapeutic equivalence in pharmaceutical products in Chile and its amendments; and Decree No. 945 of 2014 of the MoH approving Technical Guideline No. 170 on sanitary registration for biotechnological products derived from recombinant DNA techniques and its amendments, among several others).
Medical devices are governed by the Sanitary Code and the regulations set forth in Supreme Decree No. 825/1999 which contains the Regulations for Products and Devices of Medical Use. Furthermore, medical devices law and regulations incorporate a progressive implementation through grounded Supreme Decrees issued by the Ministry of Health –prior report issued by the ISP–, indicating the specific medical devices which will need to fulfill the provisions included in the Sanitary Code and Supreme Decree No. 825/99 in order to be manufactured, imported, commercialized and distributed in Chile.
Currently, regulated medical devices to which sanitary restrictions apply include latex surgical gloves for single use, latex medical examination gloves and latex condoms (Decree No. 342/2004 of the MoH), sterile hypodermic needles for single use and sterile hypodermic syringes for single use (Decree No. 1.887/2007 of the MoH) and synthetic masculine condoms and feminine condoms (Decree No. 93/2018 of the MoH).
There is no general regulatory reimbursement process or pricing laws for pharmaceutical products or medical devices.
Nevertheless, the health coverage of pharmaceutical products and medical devices is based on a public and private insurance system and universal coverage programs, being the most relevant the Explicit Health Guarantees (GES plan) and the High Cost Treatment Financial Protection System (Ley Ricarte Soto).
3. What are the steps to obtaining authorization to develop, test, and market a product?
PHARMACEUTICAL PRODUCTS
Any pharmaceutical product, whether imported or manufactured in the country, requires a sanitary registration (marketing authorization) in order to be distributed or used under any title in Chile. A pharmaceutical product may be exceptionally authorized by the ISP to be used temporarily without prior sanitary registration if an epidemic, emergency or catastrophe occurs, or if required for an urgent medical use or for scientific research or clinical trials. We will later provide more information on the provisional use of pharmaceutical products.
In general terms, for the sanitary registration of a pharmaceutical product the applicant will be required to comply with general requirements including the submission of administrative information, technical information, pharmaceutical quality information and data on safety and efficacy of the product. Special requirements will also be applicable for fixed dose combination pro-ducts, pharmaceutical combination products, phytopharmaceutical products; homeopathic products and biologicals.
Safety and efficacy data, including full preclinical and clinical studies for the product will be necessary to be submitted in order to achieve the sanitary registration of a pharmaceutical product under the standard registration procedure (procedimiento ordinario de registro), applicable, in general terms for innovator products. Nonetheless, Chilean regulations, in specific cases, also include the possibility to file for a simplified procedure (procedimiento simplificado de registro), permitting the omission of specific safety and efficacy data, available for generics products, as will be described.
Additionally, during 2020, modifications were made to Supreme Decree No. 3/2010 with the purpose of modifying the conditions for the abbreviated registration procedure and incorporating a new accelerated registration process, as will be described.
3.A. Standard Procedure (“Procedimiento Ordinario”) for the registration of Pharmaceutical Products (new drugs, biologics)
The standard procedure is the general procedure established under Chilean regulations for the sanitary registration of pharmaceutical products and will be applicable in all cases defined under article 53 of Supreme Decree No- 03/2010 which, in general, relates to:
- Pharmaceutical products incorporated for the first time in the field of medicine in Chile;
- For biological products;
- Products containing an active ingredient of a previously registered product granted with regulatory data protection;
- For products including a new therapeutic utility, posology, route of administration or age group of a previously registered product.
- For products which include a modification in the composition and concentration of the active ingredients, or present new salts, esters, isoforms or complexes for an active ingredient of a previously registered product or constitute a fixed dose combination of active ingredients which, separately, may have or not prior sanitary approval.
- Products having a different dosage form of a previously registered product, modifying the release of the active ingredient.
- For the first registration of a combination pharmaceutical product
As indicated above, full preclinical and clinical data on safety and efficacy will be required to be submitted under the standard procedure for sanitary registration of these products.
For biotechnological products, and within the standard procedure of registration, Chilean regulations allow for the abbreviation of clinical data to certify the safety and efficacy provided such product has the same active ingredient, unitary dosage, pharmaceutical form and administration route than another registered reference product. This sets forth the possibility of a biosimilar pathway for specific biotechnological products. The biosimilar pathway, however, is only available for the active ingredients and their respective presentations included in Technical Guideline No. 170 approved by Decree No. 945 of 2014, and its amendments. Technical Guideline No. 170 is based upon the WHO Guidelines for similar biotherapeutic products and structures the biosimilar pathway upon a stepwise comparability on characterization and quality, non-clinical and clinical stages, head to head with the reference product (which is also specifically set within such gui-dance). It also includes provisions in connection to pharmacovigilance and extrapolation of indications.
The administrative steps for the sanitary registration of a product under the standard procedure is described below:
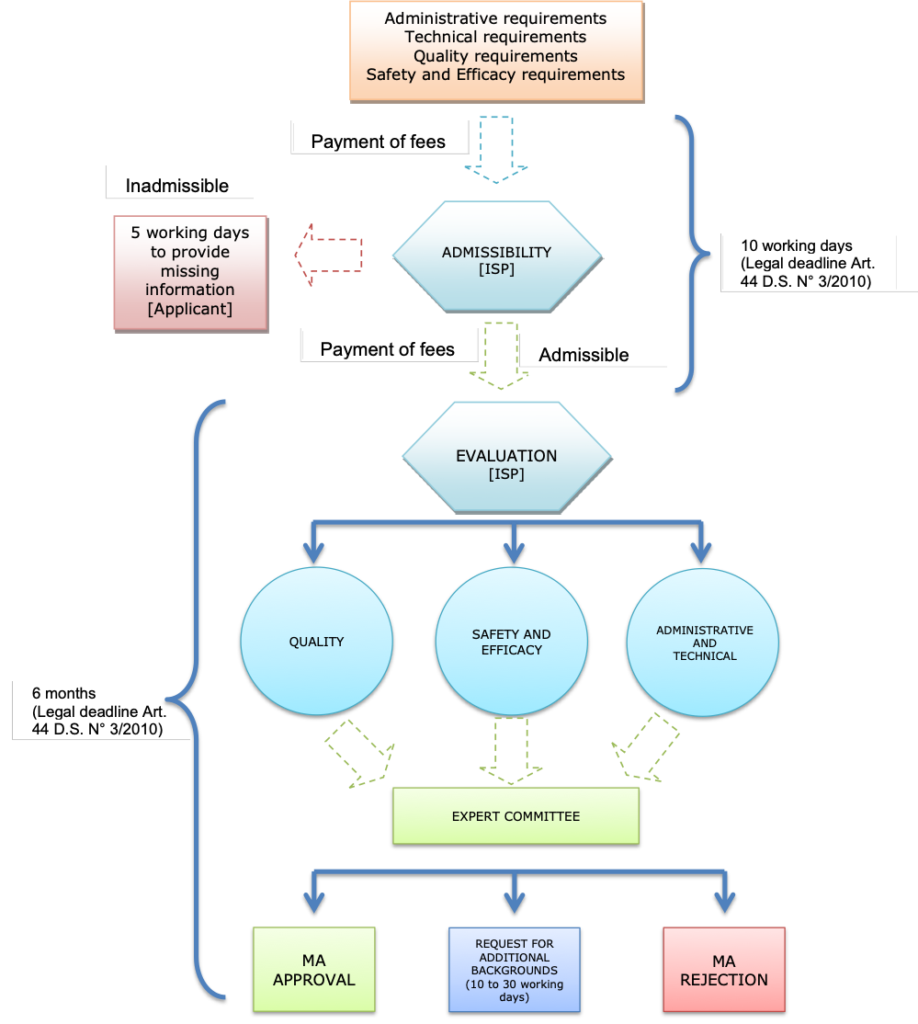
Note: MA= marketing authorization
3.B. Simplified Procedure for the registration of Pharmaceutical Products (generics)
The simplified procedure allows for the omission of specific data regarding safety and efficacy of the pharmaceutical product filed for registration and it is permitted, in general, for pharmaceutical products containing the same active ingredient, in the same pharmaceutical dosage form and the same administration route as another product which currently has or had in the past a sanitary registration granted by the ISP and which has not been cancelled for public safety issues.
Specific therapeutic equivalence studies will be required for products containing active ingredients as per Decree No. 500 of 2012 of the MoH approving Technical Guideline No. 136 which establishes the active ingredients included in pharmaceutical products that must demonstrate therapeutic equivalence and its amendments.
The administrative steps for the sanitary registration of a product under the simplified procedure is described below:
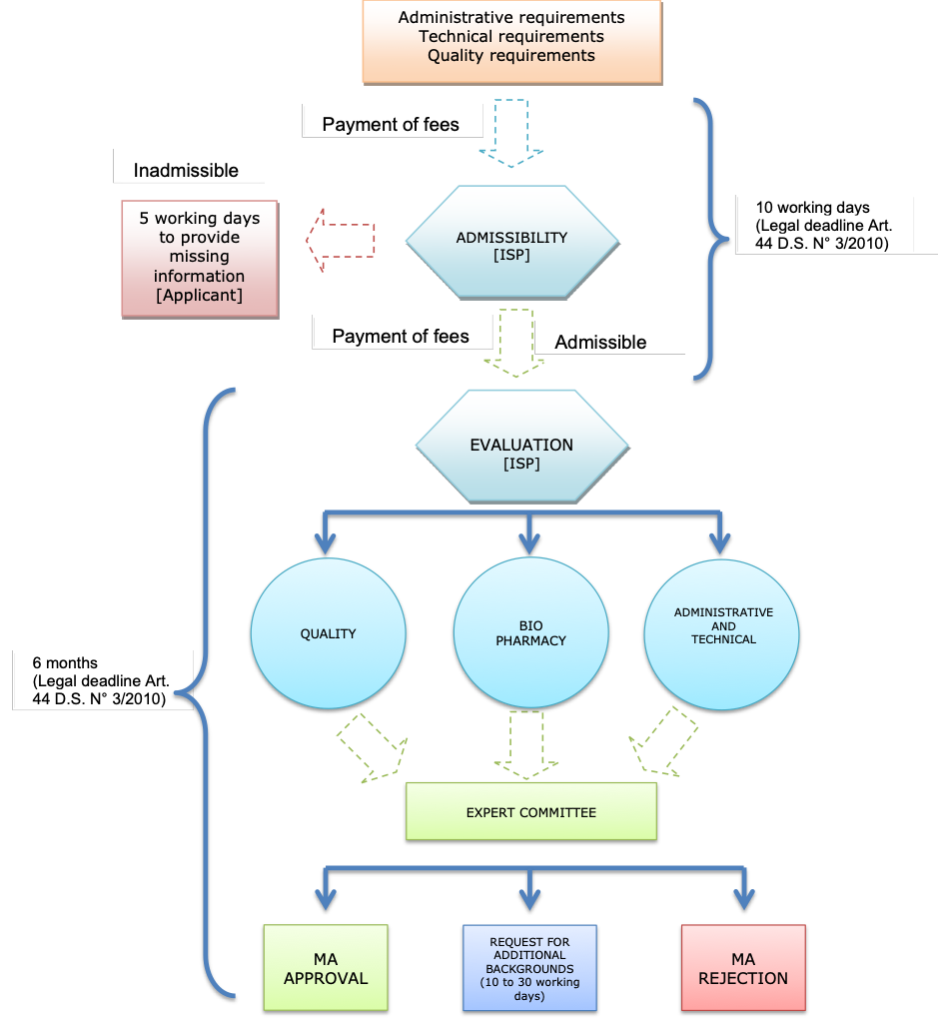
Note: MA= marketing authorization
3C. Abbreviated Procedure for the registration of Pharmaceutical Products (abbreviated pathway)
On August 21st, 2020, article 51 of Supreme Decree No. 03/2010 was modified to amend the abbreviated procedure, which now may be declared ex officio by the ISP or at the request of the interested party.
In the event that the abbreviated procedure petition is filed by the interested party, the application can be based on the following circumstances:
- When the pharmaceutical product is needed to be distributed to the population in compliance with plans or health programs approved by the Ministry of Health, in which certain situations of health risk or specific pathologies are addressed and which are destined for certain groups of people, within the framework of national public health interests.
- When the pharmaceutical product has been included in the list of products incorporated in the National Formulary of Medicines, in which case the interested party must use the monographs of the Formulary to speed up the processing of the registration.
According to the regulation, the abbreviated procedure timelines should not exceed four months.
3D. Accelerated Procedure for the registration of Pharmaceutical Products (accelerated pathway)
On August 21st, 2020, article 54 of Supreme Decree No. 03/2010 was also modified to incorporate a new accelerated registration procedure.
This accelerated procedure is based on the products’ approval by High Surveillance Regulatory Agencies, as defined in the provisions, and the approval timeline should not exceed three months since the product is declared admissible, as long as all registration requirements are fulfilled.
To request the accelerated registration procedure, the applicant must indicate in the application the existence of a sanitary registration or authorization for use granted by any of the following regulatory agencies (High Surveillance Regulatory Agencies:
I. Those defined as stringent regulatory authorities in Annex 5 of the “WHO Expert Committee on Specifications for Pharmaceutical Preparations – WHO Technical Report Series, No. 986 – Forty-eighth Report” and its subsequent modifications.
II. Those classified as Level IV in the Evaluation System of National Drug Regulatory Authorities of the Pan American Health Organization.
III. Members of the “Pharmaceutical Inspection Co-operation Scheme” (PIC/S).
In order to obtain the sanitary registration of the pharmaceutical product under the accelerated procedure, the applicant must submit the same supporting information given to the Regulatory Agency that granted the registration (reference agency), request the same therapeutical indication and submit the Certificate of Pharmaceutical Product (CPP) of the reference agency.
The review carried out by the ISP must take into account the revision already performed by the respective reference Agency.
This procedure will not be applicable in the following cases:
- Biological products, unless there is a founded resolution from the Ministry of Health that allows this exception.
- When there are public health reasons regarding a certain pharmaceutical product or category of products, which will be qualified through a founded resolution of the Ministry of Health.
- Products for which the sanitary registration has been denied in one or more High Surveillance Regulatory Agencies.
Provisional Use of Pharmaceutical Products
According to article 99 of the Sanitary Code, the ISP may provisionally authorize the distribution, sale and use of pharmaceutical products without prior registration, for clinical trials or other types of scientific research, as well as for urgent medicinal uses derived from situations of shortage or inaccessibility that may affect people considered individually or collectively.
In this sense, Decree No. 50/2019 amended article 21 of Supreme Decree No. 03/2010 to specify the causes through which the provisions of article 99 of the Sanitary Code would be applied:
- Urgent medicinal use derived from situations of shortage or inaccessibility, affecting people, considered collectively.
- Those pharmaceutical products for urgent medicinal use imported for the exclusive consumption of the importer (for a maximum treatment of 6 months).
- In the case of products to be used in scientific research or clinical trials, with a prior favorable report from the corresponding ethics committee or committees, in accordance with the rules on clinical trials performed on human beings, approved by the Ministry of Health.
In the case of letters a) and b) indicated above, they will be presented to the ISP proving, by any means, the authorization granted by the health authority of the country of origin or manufacture, as appropriate, and the prescription of the pharmaceutical product must be issued by a professional enabled to prescribe, stating the need and duration of the treatment (with a maximum of 6 months of treatment for letter b)). These authorizations may be requested by the interested party as many times as necessary.
MEDICAL DEVICES
Concerning medical devices, as of today, only the following medical devices require a sanitary registration:
- Latex surgical gloves for single use
- Latex gloves for medical examination
- Male natural, rubber and latex condoms
- Female condoms
- Sterile hypodermic needles for single use
- Sterile hypodermic syringes for single use
For regulated medical devices, the ISP will evaluate the product backgrounds based on the risk classification of the medical device.
- Class I: includes devices having a very low level of risk
- Class II: includes devices having a moderate level of risk
- Class III: includes devices having a high risk potential
- Class IV: includes devices considered the most critical in the matter of risk
According to their class, medical devices must comply with quality, safety and efficacy requirements, which will be reviewed by ISP, and certification of the verification of conformity, granted by authorized entities or by the ISP in the absence of them.
All other medical devices, are currently “non-regulated medical devices” which may be imported to Chile and commercialized in the country without further restrictions and require no sanitary registrations or authorization, although they may be subject to voluntary measures.
For regulated medical devices, the applicant must request the “registration” of the medical device before the ISP through filling in form SDM/005 as made available by the ISP, and submitting:
- Certification of the verification of conformity issued by an entity authorized by the ISP.
- Certificate for exporting purposes or free sale certificate issued by the rele-vant sanitary authority, duly legalized.
- Document issued by the manufacturer, where the applicant is recognized as the distributor of its products (mandatory for importers and distributors).
- Certificate of quality management system in the manufacturing process (e.g. ISO 13485; ISO 9001, or others).
To commercialize a regulated medical device it will be necessary to have:
- Authorization of warehouse of storage for medical devices, duly issued by the competent Health Regional Ministerial Secretariat (SEREMI).
- Customs destination certificate, issued by the relevant SEREMI which will permit the transport of the product to the authorized warehouse of the provider.
- Use and Disposition (AUD) resolution, granted by the ISP in order to authorize the use and distribution of controlled medical devices imported to Chile.
- Certification of the verification of conformity of the medical device performed by the authorized entity; and
- Sanitary registration of the medical device in the ISP (as previously explained).
4. What are the approximate fees for each authorization?
Currently, the fees for obtaining a marketing authorization are:
| GOVERMENT FEES IN USD FOR MARKETING AUTHORIZATION FOR A: | |
| NEW DRUG (Standard Procedure) | USD 2000 |
| BIOLOGICAL (Standard Procedure) | USD 2000 |
| GENERICS (Simplified Procedure) | USD 1600 |
| GOVERMENT FEES IN USD FOR MEDICAL DEVICES | |
| Registration of the company before ISP | USD 320 |
| Registration of regulated medical device (per product) | USD 100 |
| Certificate of Review of the Antecedents that accompany the non-regulated medical device (per product) | USD 330 |
| Declaration of regulatory status of medical devices (voluntary submission) | USD 25 |
5. For how long are marketing authorizations/registrations valid? How are marketing authorizations/registrations renewed?
Sanitary registrations for pharmaceutical products will last for a period of 5 years, which is renewable for equal and successive periods, if not cancelled.
6. How does the authorization process differ between brand-name products and generic products? Are there differences for local manufacturers versus foreign-owned manufacturers?
Pursuant to article 20 of Supreme Decree No. 03/2010 (S.D. No. 03/2010), every pharmaceutical product, whether imported or manufactured in the country, is required to have a sanitary registration which is provided by the ISP for its distribution or use under any title in Chile. In this regard, there are no differences for local manufacturers or foreign-owned manufacturers.
As stated in question No. 3 above, innovator products shall be subject to the Standard procedure (procedimiento ordinario de registro) and will be required to submit, among other information, the full preclinical and clinical studies to evidence safety and efficacy of the product. For biological products, the generic pathway or simplified procedure for registration is prohibited. Nevertheless, for specific biotechnological products included in Technical Guideline No. 170, a biosimilar pathway is available, permitting the reduction or abbreviation of safety and efficacy data based upon head to head, stepwise comparability studies with the reference biotechnological product in all quality-characterization, non-clinical and clinical stages.
On the other hand, our regulation sets forth the simplified procedure (procedimiento simplificado de registro), which is applicable for generic products. This pathway can be used for pharmaceutical products that have the same active ingredient, in the same amount per pharmaceutical form and the same route of administration as another product that was previously registered. The applicant in this procedure is not required to provide, with the sanitary application, the scientific information related to safety and efficacy required for the standard procedure, unless requested by the ISP by means of grounded resolutions. However, specific therapeutic equivalence studies will be required for products containing active ingredients as per Decree No. 500 of 2012 of the MoH approving Technical Guideline No. 136 establishing the active ingredients included in pharmaceutical products that must demonstrate therapeutic equivalence and its amendments.
7. How are combination products (drug + drug, drug + biologic, drug + device, biologic + device, drug + biologic + device) regulated?
A pharmaceutical combination product is defined under Chilean regulations as a product comprising two or more pharmaceutical products included in a single packaging to be administered sequentially or simultaneously.
For the registration of a pharmaceutical combination product, the applicant must submit all general applicable information as explained in question No. 3 above. Furthermore, Chilean regulations requires for the applicant to prove the safety and efficacy of the proposed combined use of the pharmaceutical products and, additionally, demonstrate:
- That each pharmaceutical product must contribute to the therapeutic effect of the combination product.
- That the dosage of each product, the administration therapeutic scheme and treatment duration must provide safety and efficacy to the combination product, without risk of increasing adverse reactions.
- Compatibility between the ingredients, including excipients, used in each pharmaceutical product from a chemical, pharmacological, pharmacokinetic and biopharmaceutical standpoint, in vitro and in vivo, as applicable.
- That secondary, collateral or toxic effects are of the same or minor intensity as individually shown for each pharmaceutical product.
Pharmaceutical combination products may not include phytopharmaceuticals or homeopathic products, combined among themselves or with other pharmaceutical products.
Combination of devices or devices with pharmaceutical products are not expressly regulated. Nevertheless, the ISP has expressed its criteria in the sense that products which combine a medical device with a pharmaceutical product having only an auxiliary function (e.g. heparin covered catheters), are governed under the regulation for medical devices. On the other hand, products which include a medical device and a pharmaceutical product as a single product (e.g. prefilled syringes) are considered and governed under the rules for pharmaceutical products.
8. How is compliance with regulation monitored and evaluated? Is the regulatory regime comparable with the U.S. Food and Drug Administration or the European Medicines Agency expectations and requirements?
Compliance with regulations is monitored and evaluated by the ISP through periodical inspections, which are previously scheduled by the authority or upon complaints or denouncements received by the authority anonymously or from third parties.
Under the Sanitary Code and applicable regulations, the ISP and other sani-tary authorities are granted with ample faculties to visit, inspect and search any public or private site, to obtain and inspect records and documentation and to impose sanitary measures, applicable upon imminent risk and nece-ssary to safeguard public health. In 2016, the ISP, through Exempt Resolution No. 1290, approved an Inspection Manual for the exertion of these faculties and has also issued internal guidelines for sanitary inspections (available at: http://www.ispch.cl/sites/default/files/manual_interno_fiscalizacion_con_medidas.pdf).
If, within the sanitary inspections performed by the ISP, a sanitary infringement is detected, the authority may open a sanctioning administrative procedure (sumario sanitario), under which it may impose sanctions including fines or other penalties as indicated in question No. 9 below.
9. What is the potential range of penalties for noncompliance?
Penalties and sanctions for non-compliance of sanitary laws and regulations may be imposed by the ISP prior carrying out the respective sanctioning procedure (sumario sanitario). The penalties which it may impose upon finding a sanitary infringement include fines ranging from USD 0.1 to USD 70,000. In case or repeated infringements, previously imposed fines can be duplicated.
Additionally, the ISP can shut down the location or facility where the infringement is committed, prohibit its operation, revoke the permits or authorizations granted, suspend distribution of products, among other sanctions included in section 174 of the Sanitary Code.
10. Is there a national healthcare system? If so, how is it administered and funded?
Yes, there is a national healthcare system mainly formed and structured under Health Services (Servicios de Salud), which are the entities responsible for the execution of actions of promotion, prevention, recovering of health and rehabilitation of patients, besides of enforcing the provisions of the Sanitary Code, when appropriate.
For the developing of the referred actions, they are decentralized state entities, granted with their own legal personality and patrimony. The Assistance Network of each service is formed by the set of public healthcare assistance facilities that are part of the service.
Health coverage is primarily based on a public and private insurance system and two universal coverage programs, which are Explicit Guarantees in Health (GES plan) and a High Cost Treatment Financial Protection System.
Members of the Chilean armed forces (army, navy, air force) have a special social security statute set forth by law No. 18.948 and Law No. 19.465, including healthcare.
The Chilean healthcare system is primarily structured by a mandatory medical coverage which is required by law. This coverage is financed by health insurance contributions paid to the providers of healthcare insurance (FONASA or the ISAPREs as it will be explained in question No. 11) on a monthly basis by certain persons such as employees (in which case the relevant employers are legally bound to withhold the relevant amounts from employees’ monthly wages), independent workers, among others. The law provides for a minimum medical coverage and the additional features depend on the health institution and the health plan chosen by each individual.
11. How does the government (or public) healthcare system function with private sector healthcare?
Under Chilean healthcare regulations, there are basically two types of providers of healthcare insurance: a public insurance, which is the National Health Fund (or “FONASA”) and private insurance, consisting of Health Insurance Institutions (or “ISAPRE”), which are legal entities specifically incorporated for these purposes and authorized by, and subject to the control of, the health authorities.
The affiliation of employees to FONASA is automatic. FONASA has 2 different modalities for health attention access:
- Institutional Attention, where the beneficiaries can access to the health attention granted by the Assistance Network belonging to the National Health Services System (public hospitals).
- Free Choice Modality, reserved to people who contribute with the 7% of their monthly salary and their family, who have the option to choose both the professional and the attention facility, within a list of public and private providers who have signed an agreement with FONASA.
On the other hand, coverage by the ISAPREs must be agreed to through the execution of an affiliation agreement. The objective of the ISAPREs is to grant health benefits to its beneficiaries, whether giving them within their own attention units, or financing them by paying the services provided by clinics, hospitals and other institutions.
However, both health systems are available to the public, and affiliates may choose at any time to join an ISAPRE or to join or return to FONASA. In fact, Chilean Constitution states that all persons “have the right to choose their health system, whether public or private” (art.19 N°9 of the Chilean Constitution). However, there are certain restrictions in connection with the medical coverage which is mandatory to provide by law when changing from FONASA to an ISAPRE or vice-versa (e.g. pre-existing conditions). In general, ISAPREs are only allowed to cease or exclude coverage when permitted by law (e.g. pre-existing conditions, plastic surgery, hospitalization for rest purposes, etc.). In contrast, FONASA cannot exclude coverage.
The amounts that are not covered by the ISAPREs must be directly paid by the relevant beneficiaries (copayment).
In addition to the coverage provided by FONASA and the ISAPREs individuals may choose to hire additional healthcare insurance which is not provided by FONASA or the ISAPREs; but instead by duly registered insurance companies.
12. Are prices of drugs and devices regulated and, if so, how?
Currently, the prices of drugs and devices are not regulated in Chile.
13. How are drugs and devices used by patients paid for? What roles do public and private payers play?
Please refer to No. 11 above.
14. Who dispenses drugs and devices to patients and how are those dispensers compensated?
Drugs will be dispensed within health entities (hospitals and clinics) or healthcare professionals directly (pharmacist) in the pharmacies. Compensation for drugs and devices will depend on the patient’s health care plan (FONASA / ISAPRE / additional private insurance), coverage under Explicit Guarantees in Health (GES plan) or High Cost Treatment Financial Protection System, if available or through out-of-pocket payment.
15. What are the professional and legal responsibilities of those who dispense drugs and devices? What role do they play in providing patient care, information, and safety?
The professionals who dispense drugs must comply with all regulations applying to reception, storage, transport, distribution and dispensing of drug products. Additionally, they must be in possession of the title of Pharmacist.
According to section 129 A of the Sanitary Code, pharmacies must be under the supervision and technical direction of pharmacist who must supervise the correct dispensing of pharmaceutical products in accordance to the terms of the respective medical prescription and to personally inform a promote rational use of medicines, responding questions which patients may have. They shall also exert a permanent supervision of sanitary and technical aspects of the site, irrespective of the responsibility over administrative operation of the site which shall oversee the pharmacy staff.
Supreme Decree No. 466 of 1984 of the MoH, which sets forth the regulations for pharmacies, wholesalers, pharmaceutical warehouses, cabinets and authorized deposits, further complements such legal responsibilities for professionals authorized for the dispensing of pharmaceutical products. In this regard, the technical director of the facilities, must verify and control the adequate dispatch of medical prescriptions assuring compliance with the conditions of sale approved for every product, assuring the correct storage conditions for pharmaceutical products are observed and training and supervising the pharmacy staff in their functions and in compliance with sanitary provisions, among others.















































